- Компании
- Takeda. О компании, буклеты, каталоги, контакты
- Olympus. О компании, буклеты, каталоги, контакты
- Boston Scientific. О компании, буклеты, каталоги, контакты
- Pentax. О компании, буклеты, каталоги, контакты
- Fujifilm & R-Farm. О компании, буклеты, каталоги, контакты
- Erbe. О компании, буклеты, каталоги, контакты
- Еще каталоги
- Мероприятия
- Информация
- Обучение
- Классификации
- Атлас
- Quiz
- Разделы
- Пациенту






")












QR-код этой страницы
Для продолжения изучения на мобильном устройстве ПРОСКАНИРУЙТЕ QR-код с помощью спец. программы или фотокамеры мобильного устройства
Случайный выбор
данная функция, случайным образом выбирает информацию для Вашего изучения,
запустите выбор нажав кнопку ниже

Обратная связь
Напишите нам
Документы и приказы: ПРИКАЗ МИНЗДРАВА РФ №1113н от 19 октября 2020 «Об утверждении Порядка сообщения субъектами обращения медицинских изделий обо всех случаях выявления побочных действий, не указанных в инструкции по применению или руководстве по эксплуатации медицинского
Анонс:
Утвержден новый порядок взаимодействия медорганизаций, производителей и РЗН по случаям неблагоприятных последствий применения медизделий

Полный текст статьи:
Текстовая версия документа
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ
РОССИЙСКОЙ ФЕДЕРАЦИИ
(МИНЗДРАВ РОССИИ)
ПРИКАЗ
19 октября 2020 №1113н
Москва
Об утверждении
Порядка сообщения субъектами обращения медицинских изделий обо всех случаях выявления побочных действий, не указанных в инструкции по применению или руководстве по эксплуатации медицинского изделия, о нежелательных реакциях при его применении, об особенностях взаимодействия медицинских изделий между собой, о фактах и об обстоятельствах, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинских изделий
В соответствии с частью 3 статьи 96 Федерального закона от 21 ноября 2011 г.
№ 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации» (Собрание законодательства Российской Федерации, 2011, № 48, ст. 6724) и подпунктом 5.2.192 Положения о Министерстве здравоохранения Российской Федерации, утвержденного постановлением Правительства Российской Федерации от 19 июня 2012 г. № 608 (Собрание законодательства Российской Федерации, 2012,
№ 20, ст. 2528), пр и к аз ы в а ю:
1. Утвердить прилагаемый Порядок сообщения субъектами обращения медицинских изделий обо всех случаях выявления побочных действий,
не указанных В инструкции по применению или руководстве по эксплуатации медицинского изделия, о нежелательных реакциях при его применении,об особенностях взаимодействия медицинских изделий между собой, о фактах и об обстоятельствах, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинских изделий.
2. Настоящий приказ вступает в силу с 1 января 2021 г. и действует до 1 января 2027
Министр М.А. Мурашко
УТВЕРЖДЕН
приказом Министерства здравоохранения Российской Федерации
от«19» октября 2020 г. № 1113н
Порядок
сообщения субъектами обращения медицинских изделий обо всех случаях выявления побочных действий, не указанных в инструкции по применению или руководстве по эксплуатации медицинского изделия, о нежелательных реакциях при его применении, об особенностях взаимодействия медицинских изделий между собой, о фактах и об обстоятельствах, создающих угрозу жизни и здоровью граждан и медицинских работников при применении
и эксплуатации медицинских изделий
1. Настоящий Порядок устанавливает правила сообщения субъектами обращения медицинских изделий обо всех случаях выявления побочных действий, не указанных в инструкции по применению или руководстве по эксплуатации медицинского изделия, о нежелательных реакциях при его применении, об особенностях взаимодействия медицинских изделий между собой, о фактах и об обстоятельствах, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинских изделий (далее - Порядок).
2. Организации, созданные в установленном порядке1 на территории
Российской Федерации, либо представительства иностранных организаций,
аккредитованные в установленном порядке2 на территории Россиискои Федерации,
либо индивидуальные предприниматели, зарегистрированные на территории Российской Федерации, либо физические лица, осуществляющие технические испытания, токсикологические исследования, клинические испытания, экспертизу качества, эффективности и безопасности медицинских изделий, их государственную регистрацию, производство, изготовление, ввоз на территорию Российской Федерации, вывоз с территории Российской Федерации, подтверждение соответствия, государственный контроль, хранение, транспортировку, реализацию, монтаж, наладку, применение, эксплуатацию, в том числе техническое обслуживание, предусмотренное норматищюй, технической и (или) эксплуатационной документацией производителя, а также ремонт, утилизацию или уничтожение (далее - субъекты обращения медицинских изделий), за исключением
1 Федеральный закон от 8 августа 2001 г. № 129-ФЗ «О государственной регистрации юридических лиц и индивидуальных предпринимателей» (Собрание законодательства Российской Федерации, 2001, № 33, ст. 3431; 2020, № 31, ст. 5048).
2 Статья 21 Федерального закона 9 июля 1999 г. № 160-ФЗ «Об иностранных инвестициях в Российской Федерации» (Собрание законодательства Российской Федерации, 1999, № 28, ст. 3493; 2014, № 19, ст. 2311).
3. Производитель медицинских изделий (его уполномоченный представитель) представляет в Службу отчет о неблагоприятном событии при применении медицинского изделия (рекомендуемый образец приведен в приложении № 2 к настоящему Порядку) (далее - отчет о неблагоприятном событии) и отчет о корректирующих действиях по безопасности медицинского изделия (рекомендуемый образец приведен в приложении № 3 к настоящему Порядку) (далее - отчет о корректирующих действиях). субъектов обращения медицинских изделий, осуществляющих деятельность на территории международного медицинского кластера или на территориях инновационных научно-технологических центров3 в течение двадцати рабочих дней со дня выявления побочных действий, не указанных в инструкции по применению или руководстве по эксплуатации медицинского изделия, нежелательных реакций при его применении, особенностей взаимодействия медицинских изделий между собой, фактов и обстоятельств, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинских изделий (далее - неблагоприятное событие), направляют в Федеральную службу по надзору в сфере здравоохранения (далее - Служба) сообщение о неблагоприятном событии при применении медицинского изделия (рекомендуемый образец приведен в приложении № 1 к настоящему Порядку).
Первоначальный отчет о неблагоприятном событии представляется в следующие сроки:
а) в случае возникновения серьезной угрозы здоровью незамедлительно, но не позднее чем через 2 календарных дня после того, как производителю медицинских изделий (его уполномоченному представителю) стало известно о наличии угрозы;
б) в случае смерти или непредвиденного серьезного ухудшения состояния здоровья пользователя незамедлительно после того, как производитель медицинских изделий (его уполномоченный представитель) установил связь между применением медицинского изделия и произошедшим событием, но не позднее чем через 1О календарных дней после того, как производителю медицинских изделий (его уполномоченному представителю) стало известно о событии;
в) в прочих случаях - незамедлительно после того, как производитель медицинских изделий (его уполномоченный представитель) установил связь между применением медицинского изделия и произошедшим событием, но не позднее чем через 30 календарных дней после того, как производителю медицинских изделий (его уполномоченному представителю) стало известно о событии.
Производитель медицинских изделий (его уполномоченный представитель) вправе выполнить корректирующие действия (до направления в Службу
3 Часть 5 статьи 38 Федерального закона от 21 ноября 2011 г. № 323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации» (Собрание законодательства Российской Федерации, 2011, № 48, ст. 6724; 2020, № 29, ст. 4516).
первоначального отчета о корректирующих действиях в экстренных случаях защиты пользователей или третьих лиц от угрозы смерти или серьезного ухудшения состояния здоровья. В этом случае первоначальный отчет о корректирующих действиях должен быть представлен в Службу не позднее чем через 2 календарных дня после выполнения производителем медицинских изделий (его уполномоченным представителем) корректирующих действий.
В случае отсутствия у производителя медицинских изделий (его уполномоченного представителя) возможности проведения расследования произошедшего неблагоприятного события он должен без промедления уведомить об этом Службу.
Производитель медицинских изделий (его уполномоченный представитель) вправе обратиться в Службу за содействием в осуществлении доступа к медицинскому изделию для определения связи применения медицинского изделия с выявленным неблагоприятным событием.
4. Медицинские организации, осуществляющие деятельность в сфере обращения медицинских изделий (далее - медицинские организации), должны в том числе информировать производителя медицинских изделий (его уполномоченного представителя) о событиях, которые имеют признаки неблагоприятного события, а также предоставлять им доступ к медицинским изделиям, с применением которых могут быть связаны неблагоприятные события.
5. Сообщения о неблагоприятном событии при применении медицинских изделий, зарегистрированных в соответствии с законодательством Российской Федерации4 и находящимся в обращении на территории Российской Федерации, за исключением территории международного медицинского кластера или на территориях инновационных научно-технологических центров, направляются в Службу субъектами обращения медицинских изделий, в том числе осуществляющими их применение (пользователями, организациями здравоохранения).
В сообщениях указывается достоверная актуальная информация, подтверждаемая соответствующими документами, копии которых прилагаются к сообщению о неблагоприятном событии.
Тип и вид неблагоприятного события, указываемых в сообщениях о неблагоприятных событиях, структурируется по видам неблагоприятных событий, нежелательных реакций при применении медицинских изделий, фактов и обстоятельств, создающих угрозу причинения вреда жизни и здоровью людей при обращении зарегистрированных медицинских изделий. Рекомендуемый справочник-
4 Постановления Правительства Российской Федераци от 27 декабря 2012 г. № 1416 «Об утверждении Правил государственной регистрации медицинских изделий» (Собрание законодательства Российской Федерации, 2013, № 1, ст. 14, 2020, № 12, ст. 1792), от 3 апреля 2020 г. № 430 «Об особенностях обращения медицинских изделий, в том числе государственной регистрации серии (партии) медицинского изделия» (Собрание законодательства Российской Федерации, 2020, № 15, ст. 2284, Официальный интернет-портал правовой информации http://www.pravo.gov.ru, 4 июня 2020 г.).
кодификатор видов неблагоприятных событий, нежелательных реакций при применении медицинских изделий, фактов и обстоятельств, создающих угрозу причинения вреда жизни и здоровью людей при обращении зарегистрированных медицинских изделий (далее Кодификатор видов неблагоприятных событий), размещается на официальном сайте Службы в информационно-телекоммуникационной сети «Интернет» (далее сеть
«Интернет»).
6. По результатам корректирующих действий по безопасности медицинского изделия производитель медицинских изделий (его уполномоченный представитель) обязан выпустить уведомление по безопасности медицинского изделия (рекомендуемый образец приведен в приложении № 4 к настоящему Порядку) (далее - уведомление по безопасности), довести его до сведения пользователей медицинских изделий посредством размещения на своем официальном сайте в сети
«Интернет» и представить в Службу для проведения анализа содержащихся в уведомлении сведений и размещения при необходимости на официальном сайте Службы в сети «Интернет».
7. Отчеты о неблагоприятных событиях не представляются в Службу в следующих случаях:
а) по каждому отдельному неблагоприятному событию из тех, что описаны в уведомлениях по безопасности и произошли после расследования неблагоприятных событий и рассылки производителем медицинских изделий (его уполномоченным представителем) таких уведомлений и проведения корректирующих действий. Вместо этого производитель медицинских изделий (его уполномоченный представитель) может согласовать со Службой возможность периодического представления сводных отчетов по указанным неблагоприятным событиям, а также их содержание и сроки представления;
б) по каждому отдельному неблагоприятному событию из числа часто происходящих и задокументированных неблагоприятных событий (обозначенных в качестве таковых в анализе рисков, связанных с медицинским изделием, о которых уже были представлены отчеты, проанализированные производителем медицинских изделий (его уполномоченным представителем) и Службой). Вместо этого допускается представлять периодические сводные отчеты. Содержание и сроки представления периодических сводных отчетов должны быть согласованы с Службой;
в) о неблагоприятных событиях, связанных с очевидными дефектами медицинских изделий, которые пользователь всегда может выявить непосредственно перед использованием медицинского изделия;
г) о неблагоприятных событиях, не приведших к серьезному ухудшению состояния здоровья или смерти из-за особенностей конструкции, защищающей от возникновения угрозы вследствие неисправности медицинского изделия;
д) об ожидаемых и предвидимых неблагоприятных событиях, удовлетворяющих одновременно всем перечисленным ниже критериям:
неблагоприятные события обозначены в эксплуатационной (сопроводительной) документации на медицинское изделие;
неблагоприятные события известны в клинической практике, их можно качественно и количественно предугадать в случае, если медицинское изделие применяется и функционирует в соответствии со своим назначением;
неблагоприятные события задокументированы в технической документации на медицинское изделие с соответствующей оценкой рисков, проведенной до того, как произошло неблагоприятное событие;
неблагоприятные события клинически допустимы с точки зрения пользы медицинского изделия для каждого отдельного пациента;
е) если риск смерти или серьезного ухудшения состояния здоровья был проанализирован и признан ничтожно малым, если ни смерти, ни серьезного ухудшения здоровья не произошло, и риск был охарактеризован и задокументирован как допустимый в рамках документации, представляемой в составе регистрационного досье при регистрации медицинского изделия.
8. Для медицинских изделий класса потенциального риска применения 3, а также медицинских изделий, имплантируемых в организм человека класса потенциального риска применения 26, производитель медицинских изделий (его уполномоченный представитель) обязан проводить мониторинг безопасности и клинической эффективности после его регистрации (далее - клинический мониторинг) и ежегодно, в течение 3 лет, представлять в Службу отчеты по клиническому мониторингу (рекомендуемый образец приведен в приложении
№ 5 к настоящему Порядку).
Отчеты о клиническом мониторинге представляются производителем медицинских изделий (его уполномоченным представителем) в Службу не позднее
1 февраля, начиная с года, следующего за годом получения регистрационного удостоверения.
9. Клинический мониторинг проводится в соответствии с планом, который должен содержать следующие сведения:
а) цели и задачи клинического мониторинга с учетом имеющихся клинических данных, специфических особенностей и факторов риска, связанных с медицинским изделием;
6) схему клинического мониторинга, в том числе обоснование методов (способов) получения и статистического анализа клинических данных, выбора исследуемой популяции, критериев включения (исключения) и минимального количества субъектов в группе исследования и, где применимо, необходимость включения в исследование групп сравнения.
10. Сообщение о неблагоприятном событии, отчет о неблагоприятном событии, отчет о корректирующих действиях, уведомление по безопасности, отчет o клиническом мониторинге направляются в электронной форме через размещенную на официальном сайте Службы в сети «Интернет» автоматизированную информационную систему Службы или через приложение Службы для мобильных устройств, или через федеральную государственную информационную систему «Единый портал государственных и муниципальных услуг (функций)» или (в случае невозможности использования сети «Интернет») на бумажном носителе.
11. В организациях, указанных в пункте 2 настоящего Порядка, назначается ответственное за мониторинг безопасности медицинских изделий должностное лицо (далее - ответственное должностное лицо).
В обязанности ответственного должностного лица, входит сбор и направление в Службу информации, указанной в пунктах 3 и 5 настоящего Порядка, а также мониторинг информационных писем Службы, размещенных на официальном сайте в сети «Интернет», и проведение мероприятий, указанных в них в части медицинских изделий, находящих в обращении в организации.
Порядок осуществления работы и обязанности ответственного должностного лица субъекта обращения медицинских изделий в части мониторинга безопасности медицинских изделий регламентируются локальными нормативными актами субъекта обращения медицинских изделий.
12.
|
Информация, указанная в сообщении о неблагоприятном событии, обрабатывается и регистрируется Службой в соответствии с Порядком осуществления мониторинга безопасности медицинских изделий, утверждаемым Министерством здравоохранения Российской Федерации5
13. Защита данных, представляемых субъектами обращения медицинских изделий в Службу в рамках настоящего Порядка, от несанкционированного доступа осуществляется в соответствии с Федеральным законом «Об информации, информационных технологиях и о защите информации» от 27 июля 2006 г. No 149-ФЗ6•
5 Подпункт 5.2.191 Положения о Министерстве здравоохранения Российской Федерации, утвержденного постановлением Правительства Российской Федерации от 19 июня 2012 г. № 608 (Собрание законодательства Российской Федерации, 2012, № 26, ст. 3526).
6 Собрание законодательства Российской Федерации, 2006, № 31, ст. 3448; 2020, № 14, ст. 2035.
![]()
Приложение № 1 к Порядку сообщения субъектами обращения медицинских изделий обо всех случаях выявления побочных действий, не указанных
в инструкции по применению или руководстве по эксплуатации медицинского изделия,
o нежелательных реакциях при его применении,
об особенностях взаимодействия медицинских изделий между собой, о фактах и об обстоятельствах, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинских изделий, утвержденному приказом
Министерства здравоохранения Российской Федерации от«19» октября 2020 г. № 1113н
Рекомендуемый образец
Сообщение о неблагоприятном событии при применении медицинского изделия
1. Сведения о субъекте обращения медицинских изделий:
а) полное и сокращенное наименование (при наличии), в том числе фирменное наименование, организационно-правовая форма юридического лица, адрес места его нахождения или для индивидуального предпринимателя - фамилия, имя и отчество (при наличии), реквизиты документа, удостоверяющего личность, адрес места жительства, а также номера телефонов и адрес электронной юридического лица или индивидуального предпринимателя (при наличии) почты
б) идентификационный номер налогоплательщика;
в) основной государственный регистрационный номер записи о создании
юридического лица или основной государственный регистрационный номер индивидуального предпринимателя;
г) вид организации (организации, созданные на территории Российской Федерации, либо представительства иностранных организаций, аккредитованные на территории Российской Федерации, либо индивидуальные предприниматели, зарегистрированные на территории Российской Федерации).
2. Сведения о медицинском изделии, в отношении которого выявлено неблагоприятное событие:
а) наименование медицинского изделия в соответствии с регистрационным
удостоверением;
б) номер и дата регистрационного удостоверения на медицинское изделие; в) номер регистрационного удостоверения в едином реестре медицинских
изделий, зарегистрированных в рамках Евразийского экономического союза (при наличии);
г) вариант исполнения или модель медицинского изделия в соответствии с регистрационным удостоверением;
д) класс потенциального риска применения;
е) код вида и наименование вида медицинского изделия в соответствии с номенклатурной классификацией медицинских изделий, утвержденной приказом Министерства здравоохранения Российской Федерации от 6 июня 2012 г. № 4н (зарегистрирован Министерством юстиции Российской Федерации 9 июля 2012 г., регистрационный № 24852)1;
ж) код вида и наименование вида медицинского изделия в соответствии с номенклатурой медицинских изделий, правила ведения которой утверждены Решением Евразийской экономической комиссии от 29 декабря 2015 г. № 1772 (при наличии);
з) позиция каталога товаров, работ, услуг для государственных и муниципальных нужд в соответствии формирования и ведения в единой информационной системе в обеспечения с Правилами сфере закупок каталога товаров, работ, услуг для обеспечения государственных и муниципальных нужд и правил использования указанного каталога, утвержденными постановлением Правительства Российской Федерации от 8 февраля 2017 г. № 1453;
и) наименование производителя медицинского изделия в соответствии с регистрационным удостоверением;
к) наименование страны производителя медицинского изделия в соответствии с регистрационным удостоверением;
л) адрес места (адреса мест) производства медицинского изделия в соответствии с регистрационным удостоверением;
м) состав и комплектация медицинского изделия (при наличии) и перечень принадлежностей (при наличии) в соответствии с регистрационным удостоверением;
н) номер серии (партии), заводской номер (по применимости);
о) количество находящихся в обращении медицинских изделий (с указанием номеров серий, партий, заводских номеров), в отношении которых выявлено неблагоприятное событие, в штуках;
п) дата производства (изготовления) медицинского изделия; р) срок годности (эксплуатации) медицинского изделия;
с) дата окончания гарантийного срока, срока эксплуатации, срока службы, установленного производителем (по применимости, при наличии);
т) полное и сокращенное наименование (при наличии), в том числе фирменное наименование, организационно-правовая форма организации, которая осуществила реализацию серии (партии, заводского номера) медицинского изделия,
1 С изменениями, внесенными приказами Министерства здравоохранения Российской Федерации от 25 сентября 2014 r. № 557н (зарегистрирован Министерством юстиции Российской Федерации 17 декабря 2014 r., регистрационный № 35201), от 7 июля 2020 r. № 686н (зарегистрирован Министерством юстиции Российской Федерации 10 августа 2020 r., регистрационный№ 59225).
2 Официальный сайт Евразийского экономического союза http://www.eaeunion.org/, 31 декабря 2015 r.
3 Собрание законодательства Российской Федерации, 2017, № 7, ст. 1084; 2020, № 28, ст. 4421.
адрес ее места нахождения, или для индивидуального предпринимателя - фамилия, имя и отчество (при наличии), адрес места жительства, данные документа, удостоверяющего личность (далее - поставщик), а также идентификационный номер налогоплательщика, основной государственный регистрационный номер записи о создании юридического лица или основной государственный регистрационный номер индивидуального предпринимателя, номер телефона и адрес электронной почты (при наличии);
у) сведения о номере реестровой записи поставщика согласно Реестру уведомлений об осуществлении деятельности в сфере обращения медицинских изделий (далее - Реестр уведомлений/;
ф) наименование и адреса помещений, в которых осуществлялось хранение серии (партии, заводского номера, модели, варианта исполнения) медицинского изделия согласно сведениям Реестра уведомлений (при наличии);
х) полное и сокращенное наименование (при наличии), в том числе фирменное наименование, организационно-правовая форма организации, которая осуществляла применение серии (партии, заводского номера) медицинского изделия, адрес ее места нахождения (с указанием адресов мест применения), или для индивидуального предпринимателя - фамилия, имя и отчество (при наличии), адрес места жительства, данные документа, удостоверяющего личность, а также идентификационный номер налогоплательщика, основной государственный регистрационный номер записи о создании юридического лица или основной государственный регистрационный номер индивидуального предпринимателя, а также номера телефонов и адрес электронной почты юридического лица или индивидуального предпринимателя (при наличии);
ц) страна, наименование субъекта Российской Федерации (в случае, если событие произошло на территории Российской Федерации), населенный пункт, адрес, где произошло неблагоприятное событие;
ч) место нахождения медицинского изделия в момент направления сообщения о неблагоприятном событии (утилизировано, перемещено в карантинную зону, передано поставщику или производителю медицинского изделия (его уполномоченному представителю), продолжает применяться, иное);
ш) данные из актов, отчетов, журналов, технического обслуживания (при наличии);
щ) количество вовлеченных в неблагоприятное событие медицинских изделий (если известно).
3. Описание неблагоприятного события:
4 Пункт 12 Правил представления уведомлений о начале осуществления отдельных видов предпринимательской деятельности и учета указанных уведомлений, утвержденных постановлением Правительства Российской Федерации от 16 mоля 2009 г. № 584 (Собрание законодательства Российской Федерации, 2009, № 30, ст. 3823).
а) дата направления сведений о неблагоприятном событии в Федеральную службу по надзору в сфере здравоохранения;
6) тип сообщения (первичное, последующее, заключительное); в) дата поступления информации о неблагоприятном событии; г) дата неблагоприятного события;
д) для имплантируемых медицинских изделий:
дата имплантации; дата эксплантации;
длительность имплантации (заполняется в случае, если известна точная дата имплантации или начала эксплуатации);
сведения из подсистемы ведения специализированных регистров пациентов по отдельным нозологиям и категориям граждан, мониторинга организации оказания высокотехнологичной медицинской помощи и санаторно-курортного лечении единой государственной информационной системы в сфере здравоохранения5, иных информационных систем, предназначенных для сбора, хранения, обработки и предоставления информации, касающейся деятельности медицинских организаций и предоставляемых ими услуг, а также из федеральной государственной информационной системы «Единая автоматизированная вертикально-интегрированная информационно-аналитическая система по проведению медико-социальной экспертизы» (при наличии у субъекта обращения медицинских изделий доступа в указанные системы);
е) регистрационный номер неблагоприятного события, присвоенный производителем (при наличии);
ж) тип и вид неблагоприятного события в соответствии со справочником- кодификатором видов неблагоприятных событий, нежелательных реакций при применении медицинских изделий, фактов и обстоятельств, создающих угрозу причинения вреда жизни и здоровью людей при обращении зарегистрированных медицинских изделий;
з) описание побочных действий медицинского изделия, не указанных в инструкции по применению или руководстве по эксплуатации, нежелательных реакций при его применении, особенностей взаимодействия медицинских изделий между собой, фактов и обстоятельств, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинских изделий;
и) пользователь медицинского изделия (врач, медицинская сестра, пациент, иное);
к) применение медицинского изделия (первичное, повторное применение медицинского изделия однократного применения, применение медицинского
5 Подпункт «е» пункта 4 Положения о единой государственной информационной системе в сфере здравоохранения, утвержденного постановлением Правительства Российской Федерации от 5 мая 2018 г. № 555 «О единой государственной информационной системе в сфере здравоохранения» (Собрание законодательства Российской Федерации, 2018 № 20, ст. 2849).
изделия многократного применения, медицинское изделие после технического обслуживания или ремонта, проблема была выявлена до применения, иное);
л) категория неблагоприятного события, связанного с применением медицинского изделия:
серьезная и (или) непредвиденная побочная реакция, не указанная в инструкции по применению или руководстве по эксплуатации медицинского изделия;
побочное явление при применении медицинского изделия; особенности взаимодействия медицинских изделий между собой; ненадлежащее качество медицинского изделия;
обстоятельства, создающие угрозу жизни и здоровью населения и медицинских работников при применении и эксплуатации медицинских изделий;
иные случаи неблагоприятного события;
м) принятые пользователем или медицинской организацией меры по устранению неблагоприятного события.
4. Данные о пострадавшем:
а) пострадавший (пациент на дому, амбулаторный пациент, стационарный пациент, медицинский персонал, посетитель, технический персонал, самостоятельное лечение, иное);
б) фамилия, имя и отчество (при наличии);
в) пол и возраст, вес, рост (при наличии сведений); г) критерий серьезности (причиненный вред);
д) исход;
е) описание проблемы пострадавшего;
ж) количество пострадавших (если известно);
з) действия и помощь, оказанная медицинской организацией
пострадавшему.
5. Дополнительная информация и документы.
Приложение № 2 к Порядку сообщения субъектами обращения медицинских изделий обо всех случаях выявления побочных действий, не указанных
в инструкции по применению или руководстве по эксплуатации медицинского изделия,
o нежелательных реакциях при его применении,
об особенностях взаимодействия медицинских изделий между собой, о фактах и об обстоятельствах, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации
медицинских изделий, утвержденному приказом
Министерства здравоохраения Российской
Федерации от «19» октября 2020 г. № 1113н
Рекомендуемый образец
Отчет о неблагоприятном событии при применении медицинского изделия
1. Сведения о субъекте обращения медицинских изделий:
а) полное и сокращенное наименование (при наличии), в том числе фирменное наименование, организационно-правовая форма юридического лица, адрес места его нахождения или для индивидуального предпринимателя - фамилия, имя и отчество (при наличии), реквизиты документа, удостоверяющего личность, адрес места жительства, а также номера телефонов и адрес электронной почты юридического лица или индивидуального предпринимателя (при наличии);
6) идентификационный номер налогоплательщика,
в) основной государственный регистрационный номер записи
o создании юридического лица или основной государственный регистрационный номер индивидуального предпринимателя;
г) вид организации (организации, созданные на территории Российской Федерации, либо представительства иностранных организаций, аккредитованные на территории Российской Федерации, либо индивидуальные предприниматели, зарегистрированные на территории Российской Федерации).
2. Сведения о медицинском изделии, в отношении которого выявлено неблагоприятное событие:
а) наименование медицинского изделия в соответствии с регистрационным удостоверением;
6) номер и дата регистрационного удостоверения на медицинское изделие; в) номер регистрационного удостоверения в едином реестре медицинских изделий, зарегистрированных в рамках Евразийского экономического союза (при наличии);
г) вариант исполнения или модель медицинского изделия в соответствии с регистрационным удостоверением;
д) класс потенциального риска применения;
е) код вида и наименование вида медицинского изделия в соответствии с номенклатурной классификацией медицинских изделий, утвержденной приказом Министерства здравоохранения Российской Федерации от 6 июня 2012 г. № 4н (зарегистрирован Министерством юстиции Российской Федерации 9 июля 2012 г., регистрационный № 24852)1;
ж) код вида и наименование вида медицинского изделия в соответствии с номенклатурой медицинских изделий, правила ведения которой утверждены Решением Евразийской экономической комиссии от 29 декабря 2015 г. № 17?2 (при наличии);
з) позиция каталога товаров, работ, услуг для государственных и муниципальных нужд в соответствии формирования и ведения в единой информационной системе в обеспечения с Правилами сфере закупок каталога товаров, работ, услуг для обеспечения государственных и муниципальных нужд и правил использования указанного каталога, утвержденными постановлением Правительства Российской Федерации от 8 февраля 2017 г. № 1453;
и) наименование производителя медицинского изделия в соответствии с регистрационным удостоверением;
к) наименование страны производителя медицинского изделия в соответствии с регистрационным удостоверением;
л) адрес места (адреса мест) производства медицинского изделия в соответствии с регистрационным удостоверением;
м) состав и комплектация медицинского изделия (при наличии) и перечень принадлежностей (при наличии) в соответствии с регистрационным удостоверением;
н) номер серии (партии), заводской номер (по применимости);
о) количество находящихся в обращении медицинских изделий (с указанием номеров серий, партий, заводских номеров), в отношении которых выявлено неблагоприятное событие, в штуках;
п) дата производства (изготовления) медицинского изделия; р) срок годности (эксплуатации) медицинского изделия;
с) дата окончания гарантийного срока, срока эксплуатации, срока службы, установленного производителем (по применимости, при наличии);
1 С изменениями, внесенными приказами Министерства здравоохранения Российской Федерации от 25 сентября 2014 г. № 557н (зарегистрирован Министерством юстиции Российской Федерации 17 декабря 2014 г., регистрационный № 35201), от 7 июля 2020 г. № 686н (зарегистрирован Министерством юстиции Российской Федерации 10 августа 2020 г., регистрационный№ 59225).
2 Официальный сайт Евразийского экономического союза http://www.eaeunion.org/, 31 декабря 2015 г.
3 Собрание законодательства Российской Федерации, 2017, № 7, ст. 1084; 2020, № 28, ст. 4421.
т) полное и сокращенное наименование (при наличии), в том числе фирменное наименование, организационно-правовая форма организации, которая осуществила реализацию серии (партии, заводского номера) медицинского изделия, адрес ее места нахождения, или для индивидуального предпринимателя-фамилия, имя и отчество (при наличии), адрес места жительства, данные документа, удостоверяющего личность (далее - поставщик), а также идентификационный номер налогоплательщика, основной государственный регистрационный номер записи о создании юридического лица или основной государственный регистрационный номер индивидуального предпринимателя, номер телефона и адрес электронной почты (при наличии);
ф) наименование и адреса помещений, в которых осуществлялось хранение серии (партии, заводского номера, модели, варианта исполнения) медицинского изделия согласно сведениям Реестра уведомлений (при наличии);у) сведения о номере реестровой записи поставщика согласно Реестру уведомлений об осуществлении деятельности в сфере обращения медицинских изделий (далее-Реестр уведомлений)4
х) полное и сокращенное наименование (при наличии), в том числе фирменное наименование, организационно-правовая форма организации, которая осуществляла применение серии (партии, заводского номера) медицинского изделия, адрес ее места нахождения (с указанием адресов мест применения), или для индивидуального предпринимателя - фамилия, имя и отчество (при наличии), адрес места жительства, данные идентификационный номер документа, удостоверяющего личность, а также налогоплательщика, основной государственный регистрационный номер записи о создании юридического лица или основной государственный регистрационный номер индивидуального предпринимателя, а также номера телефонов и адрес электронной почты юридического лица или индивидуального предпринимателя (при наличии);
ц) страна, наименование субъекта Российской Федерации (в случае, если событие произошло на территории Российской Федерации), населенный пункт, адрес, где произошло неблагоприятное событие;
ч) место нахождения медицинского изделия в момент направления сообщения о неблагоприятном событии (утилизировано, перемещено в карантинную зону, передано поставщику или производителю медицинского изделия (его уполномоченному представителю), продолжает применяться, иное);
ш) данные из актов, отчетов, журналов, технического обслуживания (при наличии);
4 Пункт 12 Правил представления уведомлений о начале осуществления отдельных видов предпринимательской деятельности и учета указанных уведомлений, утвержденных постановлением Правительства Российской Федерации от 16 июля 2009 г. № 584 (Собрание законодательства Российской Федерации, 2009, № 30, ст. 3823).
![]()
щ) количество вовлеченных в неблагоприятное событие медицинских изделий (если известно).
3. Описание неблагоприятного события:
а) дата направления сведений о неблагоприятном событии в Федеральную службу по надзору в сфере здравоохранения;
б) тип сообщения о неблагоприятном событии при применении медицинского изделия (первичное, последующее, заключительное);
в) тип отчета о неблагоприятном событии при применении медицинского изделия (первоначальный, последующий заключительный);
г) дата поступления информации о неблагоприятном событии; д) дата неблагоприятного события;
е) для имплантируемых медицинских изделий: дата имплантации; дата эксплантации; длительность имплантации (заполняется в случае, если известна точная дата имплантации или начала эксплуатации);
ж) дата отчета о неблагоприятном событии при применении медицинского изделия;сведения из подсистемы ведения специализированных регистров пациентов по отдельным нозологиям и категориям граждан, мониторинга организации оказания высокотехнологичной медицинской помощи и санаторно-курортного лечении единой государственной информационной системы в сфере здравоохранения5 иных информационных систем, предназначенных для сбора, хранения, обработки и предоставления информации, касающейся деятельности медицинских организаций и предоставляемых ими услуг, а также из федеральной государственной информационной системы «Единая автоматизированная вертикально-интегрированная информационно-аналитическая система по проведению медико-социальной экспертизы» (при наличии у субъекта обращения медицинских изделий доступа в указанные системы);
з) регистрационный номер неблагоприятного события, присвоенный производителем (при наличии);
и) тип и вид неблагоприятного события в соответствии со справочником- кодификатором видов неблагоприятных событий, нежелательных реакций при применении медицинских изделий, фактов и обстоятельств, создающих угрозу причинения вреда жизни и здоровью людей при обращении зарегистрированных медицинских изделий;
5 Подпункт «е» пункта 4 Положения о единой государственной информационной системе в сфере здравоохранения, утвержденного постановлением Правительства Российской Федерации от 5 мая 2018 г. № 555 «О единой государственной информационной системе в сфере здравоохранения» (Собрание законодательства Российской Федерации, 2018 № 20, ст. 2849).
к) описание побочных действий медицинского изделия, не указанных в инструкции по применению или руководстве по эксплуатации, нежелательных реакций при его применении, особенностей взаимодействия медицинских изделий между собой, фактов и обстоятельств, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинских изделий;
л) пользователь медицинского изделия (врач, медицинская сестра, пациент, иное);
м) применение медицинского изделия (первичное, повторное применение медицинского изделия однократного применения, применение медицинского изделия многократного применения, медицинское изделие после технического обслуживания или ремонта, проблема была выявлена до применения, иное);
н) категория неблагоприятного события, связанного с применением медицинского изделия: серьезная и (или) непредвиденная побочная реакция, не указанная в инструкции по применению или руководстве по эксплуатации медицинского изделия; побочное явление при применении медицинского изделия; особенности взаимодействия медицинских изделий между собой; ненадлежащее качество медицинского изделия; обстоятельства, создающие угрозу жизни и здоровью населения и медицинских работников при применении и эксплуатации медицинских изделий; иные случаи неблагоприятного события;
о) принятые пользователем или медицинской организацией меры по устранению неблагоприятного события.
4. Данные о пострадавшем:
а) пострадавший (пациент на дому, амбулаторный пациент, стационарный пациент, медицинский персонал, посетитель, технический самостоятельное лечение, иное);
б) фамилия, имя и отчество (при наличии);
в) пол и возраст, вес, рост (при наличии сведений); г) критерий серьезности (причиненный вред); персонал,
д) исход;
е) описание проблемы пострадавшего;
ж) количество пострадавших (если известно);
з) действия и помощь, оказанная медицинской организацией пострадавшему.
5. Дополнительная информация:
а) предварительное заключение и анализ производителя (его уполномоченного представителя) (для первоначального/последующего отчета);
б) начальные корректирующие действия, выполненные производителем;
в) предполагаемая дата следующего отчета;
г) результаты анализа заключительного расследования производителя (для заключительного отчета);
д) корректирующие действия по безопасности медицинского изделия;
е) сроки реализации корректирующих действий по безопасности медицинского изделия;
ж) количество подобных неблагоприятных событий с таким же типом медицинского изделия с подобной же причиной неблагоприятного события и видом в соответствии с Кодификатором видов неблагоприятных событий, известных производителю (его уполномоченному представителю).
![]()
Приложение № 3 к Порядку сообщения субъектами обращения медицинских изделий обо всех случаях выявления побочных действий, не указанных
в инструкции по применению или руководстве по эксплуатации медицинского изделия,
o нежелательных реакциях при его применении,
об особенностях взаимодействия медицинских изделий между собой, о фактах и об обстоятельствах, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинских изделий, утвержденному приказом
Министерства здравоох анения Российской Федерации от «19» октября 2020 г. № 1113н
Отчет о корректирующих действиях по безопасности медицинского изделия
1. Сведения о субъекте обращения медицинских изделий:
а) полное и сокращенное наименование (при наличии), в том числе фирменное наименование, организационно-правовая форма юридического лица, адрес места его нахождения или для индивидуального предпринимателя - фамилия, имя и отчество (при наличии), реквизиты документа, удостоверяющего личность, адрес места жительства, а также номера телефонов и адрес электронной почты юридического лица или индивидуального предпринимателя (при наличии);
б) идентификационный номер налогоплательщика;
в) основной государственный регистрационный номер записи о создании юридического лица или основной государственный регистрационный номер индивидуального предпринимателя;
г) вид организации (организации, созданные на территории Российской Федерации, либо представительства иностранных организаций, аккредитованные в на территории Российской Федерации, либо индивидуальные предприниматели, зарегистрированные на территории Российской Федерации).
2. Сведения о медицинском изделии, в отношении которого выявлено неблагоприятное событие:
а) наименование медицинского изделия в соответствии с регистрационным удостоверением;
б) номер и дата регистрационного удостоверения на медицинское изделие;
в) номер регистрационного удостоверения в едином реестре медицинских изделий, зарегистрированных в рамках Евразийского экономического союза (при наличии);
г) вариант исполнения или модель медицинского изделия в соответствии с регистрационным удостоверением;
д) класс потенциального риска применения;
е) код вида и наименование вида медицинского изделия в соответствии с номенклатурной классификацией медицинских изделий, утвержденной приказом Министерства здравоохранения Российской Федерации от 6 июня 2012 г. № 4н (зарегистрирован Министерством юстиции Российской Федерации 9 июля 2012 г., регистрационный № 24852)1;
ж) код вида и наименование вида медицинского изделия в соответствии с номенклатурой медицинских изделий, правила ведения которой утверждены Решением Евразийской экономической комиссии от 29 декабря 2015 г. № 1772 (при наличии);
з) позиция каталога товаров, работ, услуг для обеспечения государственных и муниципальных нужд в соответствии с Правилами формирования и ведения в единой информационной системе в сфере закупок каталога товаров, работ, услуг для обеспечения государственных и муниципальных нужд и правил использования указанного каталога, утвержденными постановлением Правительства Российской Федерации от 8 февраля 2017 г. № 1453;
и) наименование производителя медицинского изделия в соответствии с регистрационным удостоверением;
к) наименование страны производителя медицинского изделия в соответствии с регистрационным удостоверением;
л) адрес места (адреса мест) производства медицинского изделия в соответствии с регистрационным удостоверением;
м) состав и комплектация медицинского изделия (при наличии) и перечень принадлежностей (при наличии) в соответствии с регистрационным удостоверением;
н) номер серии (партии), заводской номер (по применимости);
о) количество находящихся в обращении медицинских изделий (с указанием номеров серий, партий, заводских номеров), в отношении которых выявлено неблагоприятное событие, в штуках;
п) дата производства (изготовления) медицинского изделия; р) срок годности (эксплуатации) медицинского изделия;
1 С изменениями, внесенными приказами Министерства здравоохранения Российской Федерации от 25 сентября 2014 г. № 557н (зарегистрирован Министерством юстиции Российской Федерации 17 декабря 2014 г., регистрационный № 35201), от 7 июля 2020 г. № 686н (зарегистрирован Министерством юстиции Российской Федерации 1О августа 2020 г., регистрационный № 59225).
2 Официальный сайт Евразийского экономического союза http://www.eaeunion.org/, 31 декабря 2015 г.
3 Собрание законодательства Российской Федерации, 2017, № 7, ст. 1084; 2020, № 28, ст. 4421.
с) дата окончания гарантийного срока, срока эксплуатации, срока службы, установленного производителем (по применимости, при наличии);
т) полное и сокращенное наименование (при наличии), в том числе фирменное наименование, организационно-правовая форма организации, которая осуществила реализацию серии (партии, заводского номера) медицинского изделия, адрес ее места нахождения, или для индивидуального предпринимателя - фамилия, имя и отчество (при наличии), адрес места жительства, данные документа, удостоверяющего личность (далее - поставщик), а также идентификационный номер налогоплательщика, основной государственный регистрационный номер записи о создании юридического лица или основной государственный регистрационный номер индивидуального предпринимателя, номер телефона и адрес электронной почты (при наличии);
у) сведения о номере реестровой записи поставщика согласно Реестру уведомлений об осуществлении деятельности в сфере обращения медицинских изделий (далее - Реестр уведомлений)4;
ф) наименование и адреса помещений в которых осуществлялось хранение серии (партии, заводского номера, модели, варианта исполнения) медицинского изделия согласно сведениям Реестра уведомлений (при наличии);
х) полное и сокращенное наименование (при наличии), в том числе фирменное наименование, организационно-правовая форма организации, которая осуществляла применение серии (партии, заводского номера) медицинского изделия, адрес ее места нахождения (с указанием адресов мест применения), или для индивидуального предпринимателя - фамилия, имя и отчество (при наличии), адрес места жительства, данные документа, удостоверяющего личность, а также идентификационный номер налогоплательщика, основной государственный регистрационный номер записи о создании юридического лица или основной государственный регистрационный номер индивидуального предпринимателя, а также номера телефонов и адрес электронной почты юридического лица или индивидуального предпринимателя (при наличии);
ц) страна, наименование субъекта Российской Федерации (в случае, если событие произошло на территории Российской Федерации), населенный пункт, адрес, где произошло неблагоприятное событие;
ч) место нахождения медицинского изделия в момент направления сообщения о неблагоприятном событии (утилизировано, перемещено в карантинную
4 Пункт 12 Правил представления уведомлений о начале осуществления отдельных видов предпринимательской деятельности и учета указанных уведомлений, утвержденных постановлением Правительства Российской Федерации от 16 июля 2009 г. № 584 (Собрание законодательства Российской Федерации, 2009, № 30, ст. 3823).
зону, передано поставщику или производителю медицинского изделия (его уполномоченному представителю), продолжает применяться, иное);
ш) данные из актов, отчетов, журналов технического обслуживания (при наличии);
щ) количество вовлеченных в неблагоприятное событие медицинских изделий (если известно).
3. Описание неблагоприятного события:
а) дата направления сведений о неблагоприятном событии в Федеральную службу по надзору в сфере здравоохранения;
б) тип сообщения о неблагоприятном событии при применении медицинского изделия (первичное, последующее, заключительное);
в) тип отчета о неблагоприятном событии при применении медицинского изделия (первоначальный, последующий заключительный);
г) дата поступления информации о неблагоприятном событии;
д) дата неблагоприятного события;
е) для имплантируемых медицинских изделий:
дата имплантации; дата эксплантации;
длительность имплантации (заполняется в случае, если известна точная дата имплантации или начала эксплуатации);
сведения из подсистемы ведения специализированных регистров пациентов по отдельным нозологиям и категориям граждан, мониторинга организации оказания высокотехнологичной медицинской помощи и санаторно-курортного лечении единой государственной информационной системы в сфере здравоохранения5 иных информационных систем, предназначенных для сбора, хранения, обработки и предоставления информации, касающейся деятельности медицинских организаций и предоставляемых ими услуг, а также из федеральной государственной информационной системы «Единая автоматизированная вертикально-интегрированная информационно-аналитическая система по проведению медико-социальной экспертизы» (при наличии у субъекта обращения медицинских изделий доступа в указанные системы);
ж) дата отчета о неблагоприятном событии при применении медицинского изделия;
з) регистрационный номер неблагоприятного события, присвоенный производителем (при наличии);
5 Подпункт «е» пункта 4 Положения о единой государственной информационной системе в сфере здравоохранения, утвержденного постановлением Правительства Российской Федерации от 5 мая 2018 г. № 555 «О единой государственной информационной системе в сфере здравоохранения» (Собрание законодательства Российской Федерации, 2018 № 20, ст. 2849).
и) тип и вид неблагоприятного события в соответствии со справочником- кодификатором видов неблагоприятных событий, нежелательных реакций при применении медицинских изделий, фактов и обстоятельств, создающих угрозу причинения вреда жизни и здоровью людей при обращении зарегистрированных медицинских изделий;
к) описание побочных действий медицинского изделия, не указанных в инструкции по применению или руководстве по эксплуатации, нежелательных реакций при его применении, особенностей взаимодействия медицинских изделий между собой, фактов и обстоятельств, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинских изделий;
л) пользователь медицинского изделия (врач, медицинская сестра, пациент, иное);
м) применение медицинского изделия (первичное, повторное применение медицинского изделия однократного применения, применение медицинского изделия многократного применения, медицинское изделие после технического обслуживания или ремонта, проблема была выявлена до применения, иное);
н) категория неблагоприятного события, связанного с применением медицинского изделия: серьезная и (или) непредвиденная побочная реакция, не указанная в инструкции по применению или руководстве по эксплуатации медицинского изделия; побочное явление при применении медицинского изделия; особенности взаимодействия медицинских изделий между собой; ненадлежащее качество медицинского изделия; обстоятельства, создающие угрозу жизни и здоровью населения и медицинских работников при применении и эксплуатации медицинских изделий; иные случаи неблагоприятного события;
о) принятые пользователем или медицинской организацией меры по устранению неблагоприятного события.
4. Данные о пострадавшем:
а) пострадавший (пациент на дому, амбулаторный пациент, стационарный пациент, медицинский персонал, посетитель, самостоятельное лечение, иное);
б) фамилия, имя и отчество (при наличии); технический персонал,
в) пол и возраст, вес, рост (при наличии сведений); г) критерий серьезности (причиненный вред);
д) исход;
е) описание проблемы пострадавшего;
ж) код и термин проблемы пострадавшего в связи с неблагоприятным событием, с указанием вида в соответствии с Кодификатором видов неблагоприятных событий;
з) количество пострадавших (если известно);
и) действия и помощь, оказанная медицинской организацией пострадавшему.
5. Дополнительная информация:
а) предварительное заключение и анализ производителя (его уполномоченного представителя) (для первоначального/последующего отчета);
б) начальные корректирующие действия, выполненные производителем; в) предполагаемая дата следующего отчета;
г) результаты анализа заключительного расследования производителя (для заключительного отчета);
д) тип и вид неблагоприятного события, с указанием вида в соответствии с Кодификатором видов неблагоприятных событий;
е) корректирующие действия по безопасности медицинского изделия;
ж) сроки реализации корректирующих действий по безопасности медицинского изделия;
з) количество подобных неблагоприятных событий с таким же типом медицинского изделия с подобной же причиной неблагоприятного события и видом в соответствии с Кодификатором видов неблагоприятных событий, известных производителю (его уполномоченному представителю);
и) общие сведения и причина корректирующих действий (для отчета о корректирующих действиях);
к) описание и обоснование корректирующих действий (для отчета о корректирующих действиях);
л) рекомендации для пользователей (для отчета о корректирующих действиях);
м) мероприятия и сроки реализации корректирующих действий (для отчета o корректирующих действиях).
Приложение № 4 к Порядку сообщения субъектами обращения медицинских изделий обо всех случаях выявления побочных действий, не указанных в инструкции по применению или руководстве по эксплуатации медицинского изделия, o нежелательных реакциях при его применении, об особенностях взаимодействия медицинских изделий между собой, о фактах и об обстоятельствах, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинских изделий, утвержденному приказом Министерства здравоохранения Российской Федерации
от «19» октября 2020 г. № 1113н
Рекомендуемый образец
Уведомление по безопасности медицинского изделия
1. Сведения о субъекте обращения медицинских изделий:
а) полное и сокращенное наименование (при наличии), в том числе фирменное наименование, организационно-правовая форма юридического лица, адрес места его нахождения или для индивидуального предпринимателя - фамилия, имя и отчество (при наличии), реквизиты документа, удостоверяющего личность, адрес места жительства, а также номера телефонов и адрес электронной почты юридического лица или индивидуального предпринимателя (при наличии);
б) идентификационный номер налогоплательщика;
в) основной государственный регистрационный номер записи о создании юридического лица или основной государственный регистрационный номер индивидуального предпринимателя;
г) вид организации (организации, созданные на территории Российской Федерации, либо представительства иностранных организаций, аккредитованные на территории Российской Федерации, либо индивидуальные предприниматели, зарегистрированные на территории Российской Федерации).
2. Сведения о медицинском изделии, в отношении которого выявлено неблагоприятное событие:
а) наименование медицинского изделия в соответствии с регистрационным удостоверением;
б) номер и дата регистрационного удостоверения на медицинское изделие; в) номер регистрационного удостоверения в едином реестре медицинских
изделий, зарегистрированных в рамках Евразийского экономического союза (при наличии);
г) вариант исполнения или модель медицинского изделия в соответствии с регистрационным удостоверением;
д) класс потенциального риска применения;
е) код вида и наименование вида медицинского изделия в соответствии с номенклатурной классификацией медицинских изделий, утвержденной приказом Министерства здравоохранения Российской Федерации от 6 июня 2012 г. № 4н (зарегистрирован Министерством юстиции Российской Федерации 9 июля 2012 г., регистрационный № 24852)1;
ж) код вида и наименование вида медицинского изделия в соответствии с номенклатурой медицинских изделий, правила ведения которой утверждены Решением Евразийской экономической комиссии от 29 декабря 2015 г. № 17?2 (при наличии);
з) позиция каталога товаров, работ, услуг для государственных и муниципальных нужд в соответствии формирования и ведения в единой информационной системе в обеспечения с Правилами сфере закупок каталога товаров, работ, услуг для обеспечения государственных и муниципальных нужд и правил использования указанного каталога, утвержденными постановлением Правительства Российской Федерации от 8 февраля 2017 г. № 1453;
и) наименование производителя медицинского изделия в соответствии с регистрационным удостоверением;
к) наименование страны производителя медицинского изделия в соответствии с регистрационным удостоверением;
л) адрес места (адреса мест) производства медицинского изделия в соответствии с регистрационным удостоверением;
м) состав и комплектация медицинского изделия (при наличии) и перечень принадлежностей (при наличии) в соответствии с регистрационным удостоверением;
н) номер серии (партии), заводской номер (по применимости);
о) количество находящихся в обращении медицинских изделий (с указанием номеров серий, партий, заводских номеров), в отношении которых выявлено неблагоприятное событие, в штуках;
п) дата производства (изготовления) медицинского изделия; р) срок годности (эксплуатации) медицинского изделия;
с) дата окончания гарантийного срока, срока эксплуатации, срока службы, установленного производителем (по применимости, при наличии);
1 С изменениями, внесенными приказами Министерства здравоохранения Российской Федерации от 25 сентября 2014 г. № 557н (зарегистрирован Министерством юстиции Российской Федерации 17 декабря 2014 г., регистрационный № 35201), от 7 июля 2020 г. № 686н (зарегистрирован Министерством юстиции Российской Федерации 10 августа 2020 г., регистрационный№ 59225).
2 Официальный сайт Евразийского экономического союза http://www.eaeunion.org/, 31 декабря 2015 г.
3 Собрание законодательства Российской Федерации, 2017, № 7, ст. 1084; 2020, № 28, ст. 4421.
т) количество вовлеченных в неблагоприятное событие медицинских изделий (если известно).
3. Дополнительная информация:
а) вид корректирующего мероприятия (приостановление применения медицинского изделия, замена медицинского изделия производителем (его уполномоченным представителем), возврат медицинского изделия производителю (его уполномоченному представителю), уничтожение медицинского изделия, изменение свойств и характеристик, влияющих на качество, эффективность и безопасность медицинского изделия, совершенствование его свойства и характеристики при неизменности функционального назначения и (или) принципа действия медицинского изделия, в том числе изменение инструкций по применению или руководства по эксплуатации медицинского изделия, обновление программного обеспечения, иное) (для уведомления по безопасности);
б) описание проблемы (для уведомления по безопасности);
в) описание действий, которые должен выполнить пользователь медицинского изделия (для уведомления по безопасности);
г) указание о необходимости передачи уведомления лицам, которые должны быть информированы о проблеме и (или) должны выполнять корректирующие действия (для уведомления по безопасности);
д) указание о необходимости представления производителю (уполномоченному представителю производителя) сведений о медицинских изделиях, направленных в другие организации, и передачи этим организациям уведомления (для уведомления по безопасности);
е) контактная информация для связи по конкретному уведомлению по безопасности (наименование организации, почтовый адрес, телефон, адрес электронной почты).
Приложение № 5 к Порядку сообщения субъектами обращения медицинских изделий обо всех случаях выявления побочных действий, не указанных
в инструкции по применению или руководстве по эксплуатации медицинского изделия,
o нежелательных реакциях при его применении,
об особенностях взаимодействия медицинских изделий между собой, о фактах и об обстоятельствах, создающих угрозу жизни и здоровью граждан и медицинских работников при применении и эксплуатации медицинских изделий, утвержденному приказом
Министерства здравоохранения Российской Федерации от «19» октября 2020 г. № 1113н
Рекомендуемый образец
Отчет по клиническому мониторингу медицинского изделия
1. Сведения о субъекте обращения медицинских изделий:
а) полное и сокращенное наименование (при наличии), в том числе фирменное наименование, организационно-правовая форма юридического лица, адрес места его нахождения или для индивидуального предпринимателя - фамилия, имя и отчество (при наличии), реквизиты документа, удостоверяющего личность, адрес места жительства, а также номера телефонов и адрес электронной почты юридического лица или индивидуального предпринимателя (при наличии);
б) идентификационный номер налогоплательщика;
в) основной государственный регистрационный номер записи о создании юридического лица или основной государственный регистрационный номер индивидуального предпринимателя;
г) вид организации (организации, созданные на территории Российской Федерации, либо представительства иностранных организаций, аккредитованные на территории Российской Федерации, либо индивидуальные предприниматели, зарегистрированные на территории Российской Федерации).
2. Сведения о медицинском изделии, в отношении которого выявлено неблагоприятное событие:
а) наименование медицинского изделия в соответствии с регистрационным удостоверением;
б) номер и дата регистрационного удостоверения на медицинское изделие; в) номер регистрационного удостоверения в едином реестре медицинских
изделий, зарегистрированных в рамках Евразийского экономического союза (при наличии);
г) вариант исполнения или модель медицинского изделия в соответствии с регистрационным удостоверением;
д) класс потенциального риска применения;
е) код вида и наименование вида медицинского изделия в соответствии с номенклатурной классификацией медицинских изделий, утвержденной приказом Министерства здравоохранения Российской Федерации от 6 июня 2012 г. № 4н (зарегистрирован Министерством юстиции Российской Федерации 9 июля 2012 г., регистрационный № 24852)1
ж) код вида и наименование вида медицинского изделия в соответствии с номенклатурой медицинских изделий, правила ведения которой утверждены Решением Евразийской экономической комиссии от 29 декабря 2015 г. № 1772 (при наличии);
з) позиция каталога товаров, работ, услуг для государственных и муниципальных нужд в соответствии формирования и ведения в единой информационной системе в обеспечения с Правилами сфере закупок каталога товаров, работ, услуг для обеспечения государственных и муниципальных нужд и правил использования указанного каталога, утвержденными постановлением Правительства Российской Федерации от 8 февраля 2017 г. № 1453;
и) наименование производителя медицинского изделия в соответствии с регистрационным удостоверением;
к) наименование страны производителя медицинского изделия в соответствии с регистрационным удостоверением;
л) адрес места (адреса мест) производства медицинского изделия в соответствии с регистрационным удостоверением;
м) состав и комплектация медицинского изделия (при наличии) и перечень принадлежностей (при наличии) в соответствии с регистрационным удостоверением;
н) номер серии (партии), заводской номер (по применимости);
о) количество находящихся в обращении медицинских изделий (с указанием номеров серий, партий, заводских номеров), в отношении которых выявлено неблагоприятное событие, в штуках;
п) дата производства (изготовления) медицинского изделия; р) срок годности (эксплуатации) медицинского изделия;
с) дата окончания гарантийного срока, срока эксплуатации, срока службы, установленного производителем (по применимости, при наличии);
т) полное и сокращенное наименование (при наличии), в том числе фирменное наименование, организационно-правовая форма организации, которая осуществила реализацию серии (партии, заводского номера) медицинского изделия, адрес ее места нахождения, или для индивидуального предпринимателя - фамилия,
1 С изменениями, внесенными приказом Министерства здравоохранения Российской Федерации от 25 сентября 2014 г.
№ 557н «О внесении изменения в приложение N 1 к приказу Министерства здравоохранения Российской Федерации от 6 июня 2012 г. № 4н «Об утверждении номенклатурной классификации медицинских изделий» (зарегистрировано Министерством юстиции Российской Федерации 17 декабря 2014 г., регистрационный№ 35201).
2 Официальный сайт Евразийского экономического союза http://www.eaeunion.org/, 31 декабря 2015 г.
3 Собрание законодательства Российской Федерации, 2017, № 7, ст. 1084; 2020, № 28, ст. 4421.
имя и отчество (при наличии), адрес места жительства, данные документа, удостоверяющего личность (далее - поставщик), а также идентификационный номер налогоплательщика, основной государственный регистрационный номер записи о создании юридического лица или основной государственный регистрационный номер индивидуального предпринимателя, номер телефона и адрес электронной почты (при наличии);
у) сведения о номере реестровой записи поставщика согласно Реестру уведомлений об осуществлении деятельности в сфере обращения медицинских изделий (далее -Реестр уведомлений)4
ф) наименование и адреса помещений в которых осуществлялось хранение серии (партии, заводского номера, модели, варианта исполнения) медицинского изделия согласно сведениям Реестра уведомлений (при наличии);
х) полное и сокращенное наименование (при наличии), в том числе фирменное наименование, организационно-правовая форма организации, которая осуществляла применение серии (партии, заводского номера) медицинского изделия, адрес ее места нахождения (с указанием адресов мест применения), или для индивидуального предпринимателя - фамилия, имя и отчество (при наличии), адрес места жительства, данные документа, удостоверяющего личность, а также идентификационный номер налогоплательщика, основной государственный регистрационный номер записи о создании юридического лица или основной государственный регистрационный номер индивидуального предпринимателя, а также номера телефонов и адрес электронной почты юридического лица или индивидуального предпринимателя (при наличии).
3. Описание неблагоприятного события:
а) дата направления сведений о неблагоприятном событии в Федеральную службу по надзору в сфере здравоохранения;
б) тип сообщения о неблагоприятном событии при применении медицинского изделия (первичное, последующее, заключительное);
в) тип отчета о неблагоприятном событии при применении медицинского изделия (первоначальный, последующий заключительный);
г) для имплантируемых медицинских изделий:
дата имплантации; дата эксплантации;
длительность имплантации (заполняется в случае, если известна точная дата имплантации или начала эксплуатации);
сведения из подсистемы ведения специализированных регистров пациентов по отдельным нозологиям и категориям граждан, мониторинга организации оказания
4 Пункт 12 Правил представления уведомлений о начале осуществления отдельных видов предпринимательской деятельности и учета указанных уведомлений, утвержденных постановлением Правительства Российской Федерации от 16 июля 2009 г. № 584 (Собрание законодательства Российской Федерации, 2009, № 30, ст. 3823)
Высокотехнологичной медицинской помощи и санаторно-курортного лечении, иных информационных систем, предназначенных для сбора, хранения, обработки и предоставления информации, касающейся деятельности медицинских организаций и предоставляемых ими услуг, а также из федеральной государственной информационной системы «Единая автоматизированная вертикально-интегрированная информационно-аналитическая система по проведению медико-социальной экспертизы» (при наличии у субъекта обращения медицинского изделия доступа в указанные системы);
д) дата отчета о неблагоприятном событии при применении медицинского изделия.
4. Дополнительная информация:
а) перечень идентифицированных остаточных рисков, связанных с медицинским изделием;
б) цели и задачи клинического мониторинга безопасности и эффективности медицинского изделия;
в) схема клинического мониторинга безопасности и эффективности медицинского изделия;
г) клинические данные, полученные за отчетный период;
д) оценка клинических данных, полученных за отчетный период;
е) оценка всех клинических данных, полученных в период клинического мониторинга безопасности и эффективности медицинского изделия;
ж) заключение о необходимости (отсутствии необходимости) корректировки плана клинического мониторинга безопасности и эффективности медицинского изделия;
з) заключение о необходимости (отсутствии необходимости) выполнения корректирующих действий по безопасности медицинского изделия;
и) описание корректирующих действий по безопасности медицинского изделия (при наличии);
к) заключение (обоснование) о клинической безопасности и эффективности медицинского изделия;
л) предложение о необходимости (отсутствии необходимости) продления цикла клинического мониторинга безопасности и эффективности медицинского изделия;
м) дополнительные комментарии (при наличии).
.
Открыт общий доступ к полному тексту статьи
Рекомендуемые статьи

.jpg)


.png)
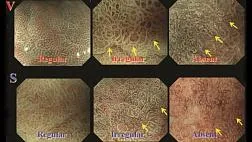
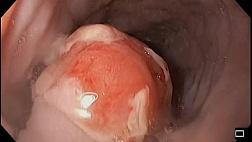
При эндоскопическом исследовании в случае бронхоэктазов в стадии ремиссии выявляется
частично диффузный бронхит I степени воспаления






Присоединяйтесь к нам в социальных сетях





Работаем и учимся при поддержке

Партнеры


Добро пожаловать на информационно-образовательный
медицинский портал EndoExpert.ru
Вы находитесь в разделе предназначенном только для специалистов (раздел для пациентов по ссылке). Пожалуйста, внимательно прочитайте полные условия использования и подтвердите, что Вы являетесь медицинским или фармацевтическим работником или студентом медицинского образовательного учреждения и подтверждаете своё понимание и согласие с тем, что применение рецептурных препаратов, обращение за той или иной медицинской услугой, равно как и ее выполнение, использование медицинских изделий, выбор метода профилактики, диагностики, лечения, медицинской реабилитации, равно как и их применение, возможны только после предварительной консультации со специалистом. Мы используем файлы cookie, чтобы предложить Вам лучший опыт взаимодействия. Файлы cookie позволяют адаптировать веб-сайты к вашим интересам и предпочтениям.
Я прочитал и настоящим принимаю вышеизложенное, хочу продолжить ознакомление с размещенной на данном сайте информацией для специалистов.
Комментарии